WARNING: HEPATIC DECOMPENSATION AND FAILURE IN PRIMARY BILIARY CHOLANGITIS PATIENTS WITH CIRRHOSIS
-
Hepatic decompensation and failure, sometimes fatal or resulting in liver transplant, have been reported with OCALIVA treatment in primary biliary cholangitis (PBC) patients with either compensated or decompensated cirrhosis.
-
OCALIVA is contraindicated in PBC patients with decompensated cirrhosis, a prior decompensation event, or with compensated cirrhosis who have evidence of portal hypertension.
-
Permanently discontinue OCALIVA in patients who develop laboratory or clinical evidence of hepatic decompensation; have compensated cirrhosis and develop evidence of portal hypertension; or experience clinically significant hepatic adverse reactions while on treatment.
INDICATION
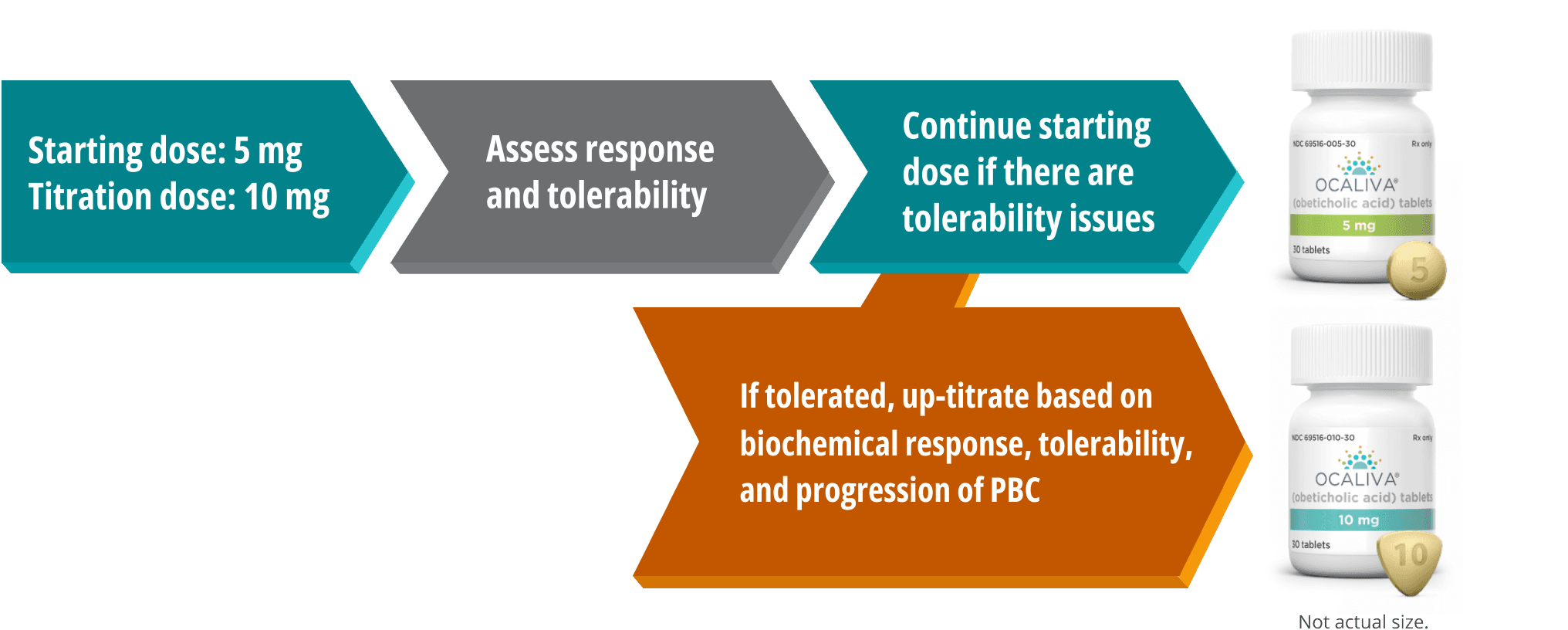
OCALIVA, a farnesoid X receptor (FXR) agonist, is indicated for the treatment of adult patients with primary biliary cholangitis (PBC)
-
without cirrhosis or
-
with compensated cirrhosis who do not have evidence of portal hypertension,
either in combination with ursodeoxycholic acid (UDCA) with an inadequate response to UDCA or as monotherapy in patients unable to tolerate UDCA.
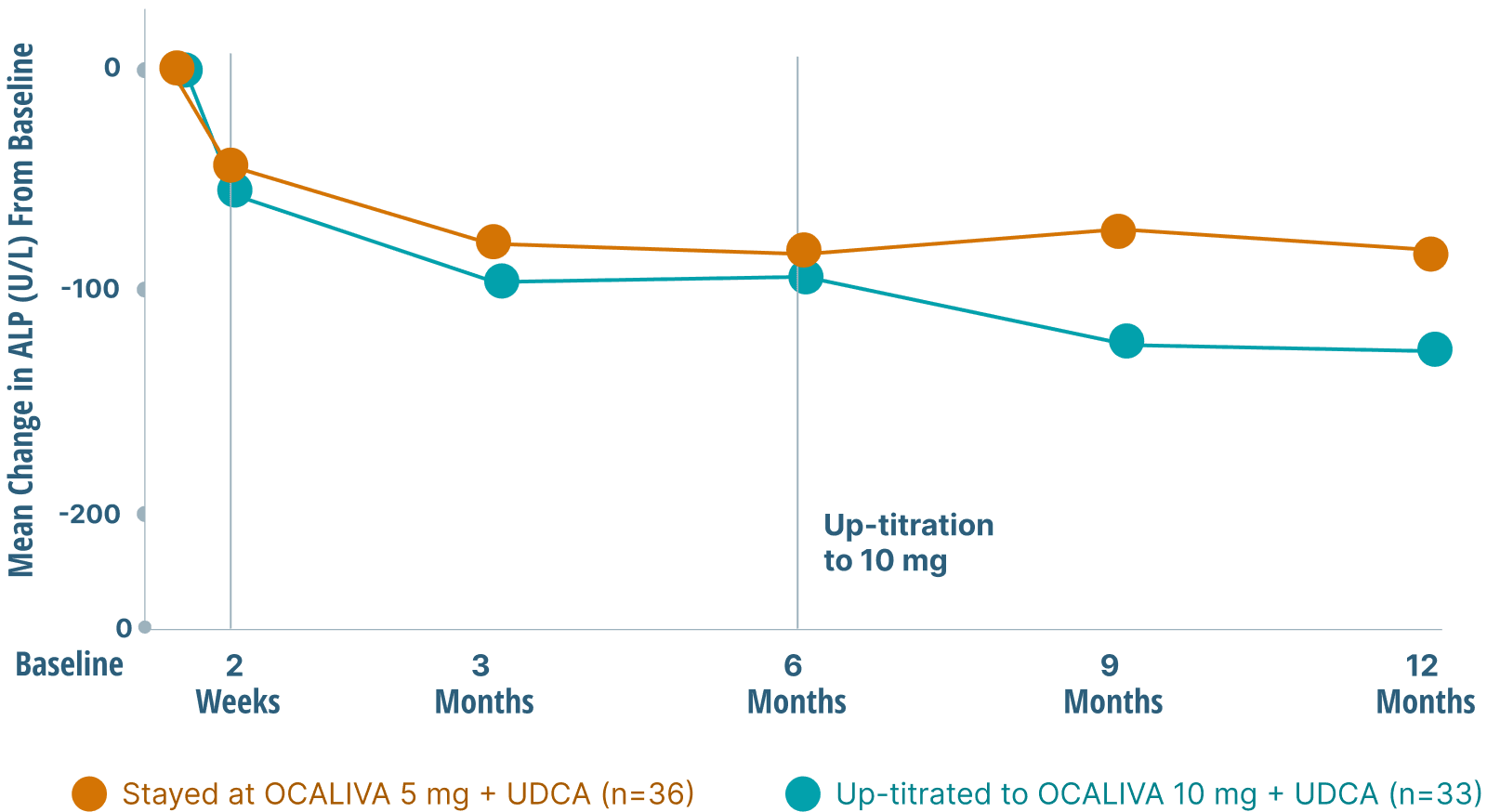
This indication is approved under accelerated approval based on a reduction in alkaline phosphatase (ALP). An improvement in survival or disease-related symptoms has not been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
Contraindications
OCALIVA is contraindicated in patients with:
-
decompensated cirrhosis (e.g., Child-Pugh Class B or C) or a prior decompensation event.
-
compensated cirrhosis who have evidence of portal hypertension (e.g., ascites, gastroesophageal varices, persistent thrombocytopenia).
-
complete biliary obstruction.
Warnings and Precautions
Hepatic Decompensation and Failure in PBC Patients with Cirrhosis
Severe Pruritus
Severe pruritus was reported in 23% of patients in the OCALIVA 10 mg arm, 19% of patients in the OCALIVA titration arm, and 7% of patients in the placebo arm in a 12-month double-blind randomized controlled clinical trial of 216 patients. Severe pruritus was defined as intense or widespread itching, interfering with activities of daily living, or causing severe sleep disturbance, or intolerable discomfort, and typically requiring medical interventions. Consider clinical evaluation of patients with new onset or worsening severe pruritus. Management strategies include the addition of bile acid binding resins or antihistamines, OCALIVA dosage reduction, and/or temporary interruption of OCALIVA dosing.
Reduction in HDL-C
Patients with PBC generally exhibit hyperlipidemia characterized by a significant elevation in total cholesterol primarily due to increased levels of high-density lipoprotein-cholesterol (HDL-C). Dose-dependent reductions from baseline in mean HDL-C levels were observed at 2 weeks in OCALIVA-treated patients, 20% and 9% in the 10 mg and titration arms, respectively, compared to 2% in the placebo arm. Monitor patients for changes in serum lipid levels during treatment. For patients who do not respond to OCALIVA after 1 year at the highest recommended dosage that can be tolerated (maximum of 10 mg once daily), and who experience a reduction in HDL-C, weigh the potential risks against the benefits of continuing treatment.
Adverse Reactions
The most common adverse reactions (≥5%) are: pruritus, fatigue, abdominal pain and discomfort, rash, oropharyngeal pain, dizziness, constipation, arthralgia, thyroid function abnormality, and eczema.
Drug Interactions
-
Bile Acid Binding Resins
Bile acid binding resins such as cholestyramine, colestipol, or colesevelam adsorb and reduce bile acid absorption and may reduce the absorption, systemic exposure, and efficacy of OCALIVA. If taking a bile acid binding resin, take OCALIVA at least 4 hours before or 4 hours after taking the bile acid binding resin, or at as great an interval as possible.
-
Warfarin
The International Normalized Ratio (INR) decreased following coadministration of warfarin and OCALIVA. Monitor INR and adjust the dose of warfarin, as needed, to maintain the target INR range when co-administering OCALIVA and warfarin.
-
CYP1A2 Substrates with Narrow Therapeutic Index
Obeticholic acid may increase the exposure to concomitant drugs that are CYP1A2 substrates. Therapeutic monitoring of CYP1A2 substrates with a narrow therapeutic index (e.g., theophylline and tizanidine) is recommended when co-administered with OCALIVA.
-
Inhibitors of Bile Salt Efflux Pump
Avoid concomitant use of inhibitors of the bile salt efflux pump (BSEP) such as cyclosporine. Concomitant medications that inhibit canalicular membrane bile acid transporters such as the BSEP may exacerbate accumulation of conjugated bile salts including taurine conjugate of obeticholic acid in the liver and result in clinical symptoms. If concomitant use is deemed necessary, monitor serum transaminases and bilirubin.
If you have questions or would like more information about OCALIVA, contact Intercept Medical Information.