For adults with primary biliary cholangitis (PBC)1
Established Safety Profile
OCALIVA has been approved since 20161
OCALIVA has a well-established safety profile, studied in a total of 432 patients with PBC in 3 placebo-controlled trials.1
MOST COMMON ADVERSE REACTIONS IN POISE 12-MONTH TRIAL (N=216)1,a,b
| Adverse Reaction | OCALIVA + UDCA | Placebo + UDCA Group (n=73) | |
|---|---|---|---|
| 5 mg →10 mg Titration Group (n=70)c | 10 mg Group (n=73) | ||
| Pruritusd | 56% | 70% | 38% |
| Fatiguee | 19% | 25% | 15% |
| Abdominal pain and discomfortf | 19% | 10% | 14% |
| Rashg | 7% | 10% | 8% |
| Arthralgia | 6% | 10% | 4% |
| Oropharyngeal pain | 7% | 8% | 1% |
| Dizzinessh | 7% | 7% | 5% |
| Constipation | 7% | 7% | 5% |
| Peripheral edema | 3% | 7% | 3% |
| Palpitations | 3% | 7% | 1% |
| Pyrexia | 0% | 7% | 1% |
| Thyroid function abnormalityi | 6% | 4% | 3% |
| Eczema | 6% | 3% | 0% |
The 10 mg arm studied in POISE is not representative of the current recommended dose schedule for OCALIVA.
aA total of 16 patients (7%) who were intolerant did not receive concomitant UDCA: 6 patients (8%) in the OCALIVA 10 mg arm, 5 patients (7%) in the OCALIVA 5 mg→10 mg titration arm, and 5 patients (7%) in the placebo arm.1
bOccurring in ≥5% of patients in either OCALIVA treatment arm and at an incidence ≥1% higher than in the placebo treatment arm.1
cPatients randomized to OCALIVA titration received OCALIVA 5 mg for the initial 6-month period. At month 6, patients who did not achieve the composite endpoint and did not have evidence of tolerability issues were titrated from 5 mg to 10 mg for the final 6 months of the trial.1
dIncludes skin eruptions, prurigo, pruritus, generalized pruritus, eye pruritus, ear pruritus, anal pruritus, vulvovaginal pruritus, and pruritic rash.1
eIncludes fatigue, tiredness, and asthenia.1
fIncludes upper abdominal pain, abdominal pain, abdominal discomfort, lower abdominal pain, abdominal tenderness, and gastrointestinal pain.1
gIncludes urticaria, rash, macular rash, papular rash, macro-papular rash, heat rash, and urticaria cholinergic.1
hIncludes dizziness, syncope, and presyncope.1
iIncludes decreased free thyroxine, increased thyroid stimulating hormone (blood), and hypothyroidism.1
97% of patients who completed the double-blind phase chose to continue OCALIVA
treatment in the long-term OLE2
Adverse events
Discontinuations
Prevalence and severity of pruritus
- Sixty-three percent of patients who entered the POISE open-label extension study had a history of PBC-related pruritus (at baseline, 56% had pruritus)4
- Throughout treatment, more pruritus days were mild than moderate or severe3
Frequency and Severity of Pruritus
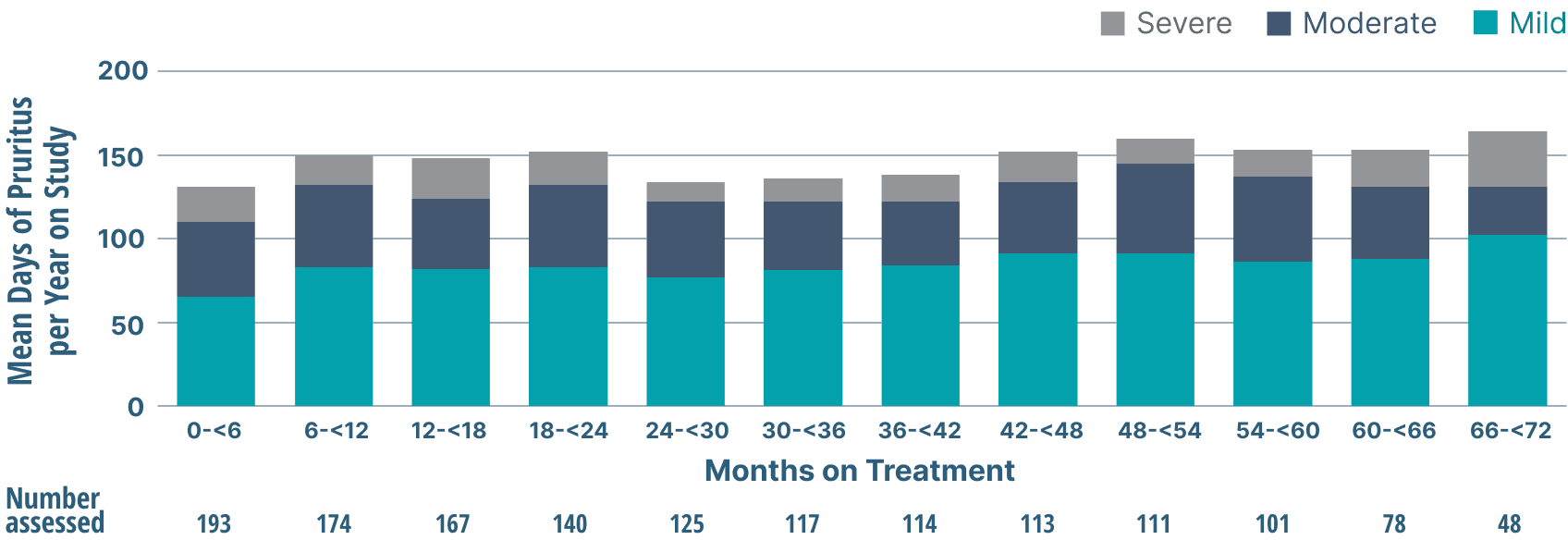
Study limitations: A maximum dose of OCALIVA 10 mg is presented, excluding data points as subjects titrated to greater than 10 mg daily.5 As this was a post hoc analysis, these results are exploratory, and no clinical conclusions should be made.
2% (4/193) discontinuation rate due to pruritus in the POISE OLE3
OCALIVA Drug-to-Drug Interactions1
| Interaction | Recommendation |
|---|---|
| Bile acid binding resins (eg, cholestryamine, colestipol, or colesevelam) adsorb and reduce bile acid absorption and may reduce the absorption, systemic exposure, and efficacy of OCALIVA | If taking bile acid binding resins, instruct patients to take OCALIVA at least 4 hours before or 4 hours after (or at as great an interval as possible) taking a bile acid binding resin |
| International normalized ratio (INR) is decreased following co-administration of warfarin and OCALIVA | Monitor INR and adjust the dosage of warfarin, as needed, to maintain target INR range when co-administering OCALIVA and warfarin |
| Obeticholic acid may increase exposure to concomitant drugs that are CYP1A2 substrates with a narrow therapeutic index (eg, theophylline and tizanidine) | Therapeutic monitoring of CYP1A2 substrates with a narrow therapeutic index (eg, theophylline and tizanidine) is recommended when co-administered with OCALIVA |
| Inhibitors of bile salt efflux pump (BSEP) may exacerbate accumulation of conjugated bile salts including taurine conjugate of obeticholic acid in the liver and result in clinical symptoms | Avoid concomitant use of inhibitors of BSEP, such as cyclosporine. If concomitant use is deemed necessary, monitor serum transaminases and bilirubin |
Routinely monitor patients for biochemical response, tolerability, and progression of PBC, including hepatic adverse reactions, during treatment.1
HDL-C, high-density lipoprotein cholesterol; OLE, open-label extension; UDCA, ursodeoxycholic acid.
References
- OCALIVA full prescribing information. Morristown, NJ: Intercept Pharmaceuticals, Inc; 2022.
- Nevens F, Andreone P, Mazzella G, et al; for the POISE Study Group. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med. 2016;375(7):631-643. doi:10.1056/NEJMoa1509840
- Data on file: INT-PB-MED-00009.
- Trauner M, Nevens F, Shiffman M, et al. Long-term efficacy and safety of obeticholic acid for patients with primary biliary cholangitis: 3-year results of an international open-label extension study. Lancet Gastroenterol Hepatol. 2019;4(6):445-453. doi:10.1016/S2468-1253(19)30094-9
- Onofrio FQ, Hirschfield GM, Gulamhusein AF. A practical review of primary biliary cholangitis for the gastroenterologist. Gastroenterol Hepatol. 2019;15(3):145-154.