For adults with primary biliary cholangitis (PBC)1
12-MONTH CLINICAL STUDY
Nearly 5x more patients who added on OCALIVA achieved reductions
in disease activity* vs UDCA alone1,5
Patients Achieving the Primary Composite Endpoint at Month 121,5,a

*Reduction in disease activity is defined as the primary composite endpoint1,5:
- ALP <1.67x ULN
- ALP decrease of ≥15%
- Total bilirubin ≤ULN
77% of patients taking OCALIVA + UDCA reduced ALP by ≥15% at 12 months vs 29%
on UDCA alone (P<0.001)1,5,a
Patients taking OCALIVA were 2.5x more likely to achieve a reduction in ALP ≥15%1
a16 patients (7%) were intolerant and did not receive concomitant UDCA: 6 patients (8%) in the OCALIVA 10 mg arm, 5 patients (7%) in the OCALIVA 5 mg→10 mg titration arm, and 5 patients (7%) in the placebo arm.1
Rapid and sustained ALP reductions over 1 year
Both OCALIVA treatment groups achieved a >30% reduction in mean ALP levels from baseline at 12 months vs an ~5% reduction with UDCA alone5,6,a
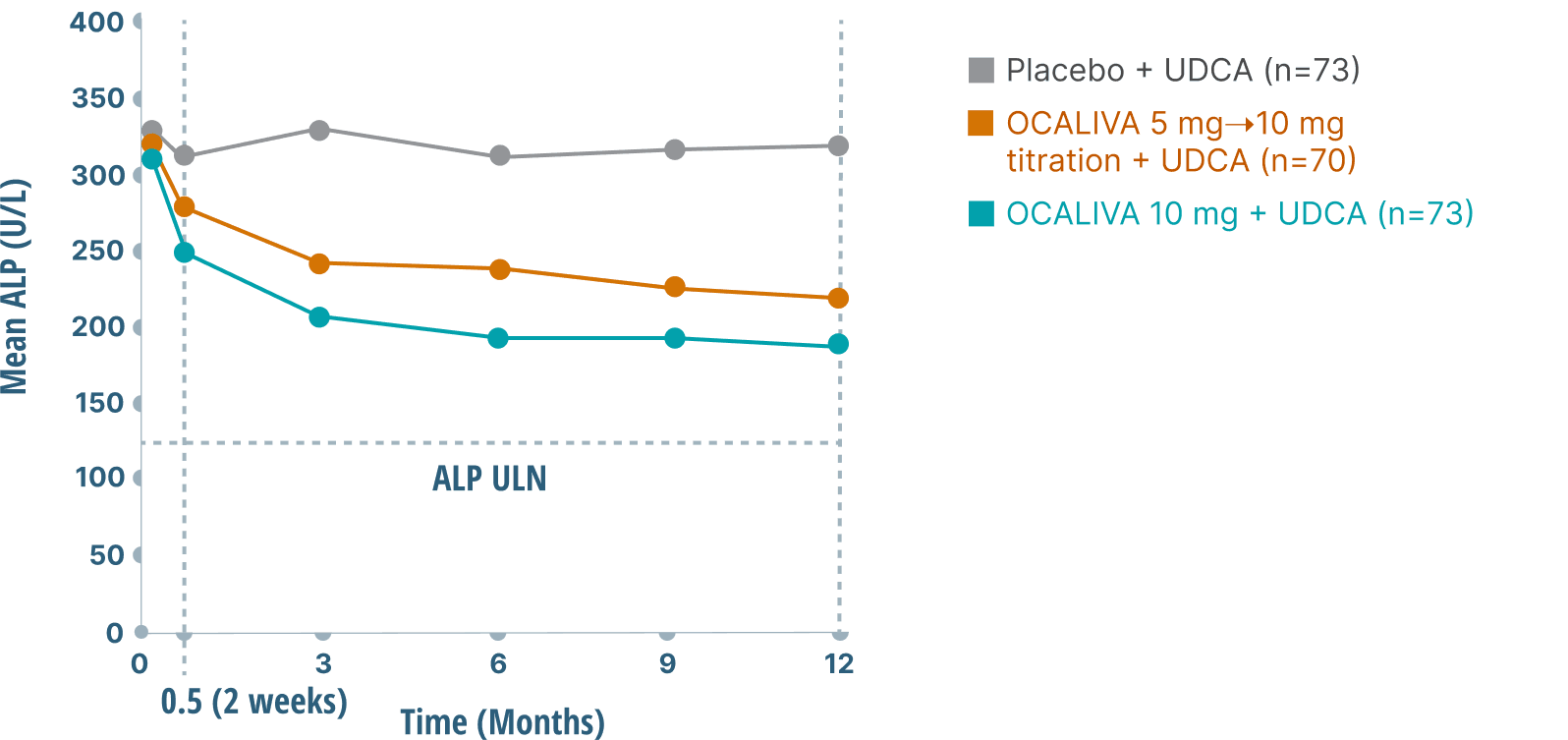
a16 patients (7%) were intolerant and did not receive concomitant UDCA: 6 patients (8%) in the OCALIVA 10 mg arm, 5 patients (7%) in the OCALIVA 5 mg→10 mg titration arm, and 5 patients (7%) in the placebo arm.1
Decreased biochemical markers at 1 year in the POISE placebo-controlled trial1,5
Secondary endpoints in POISE included change from baseline in various biochemical markers.5,a
| △ from Baseline (LS Mean ± SE) |
OCALIVA 5-10 mg (n=70) |
Placebo (n=73) |
Difference |
|---|---|---|---|
| ALP (U/L) | -112.5 ± 14.4 | -14.4 ± 14.7 | -98.1 ± 13.1 |
| GGT (U/L) | -140.8 ± 24.7 | 6.7 ± 25.6 | -147.5 ± 23.1 |
| ALT (U/L) | -21.3 ± 3.3 | -5 ± 3.3 | -16.3 ± 3.0 |
| AST (U/L) | -13 ± 4.2 | 1 ± 4.2 | -14.1 ± 3.8 |
| △ from Baseline (LS Mean ± SE) |
OCALIVA 10 mg (n=73) |
Placebo (n=73) |
Difference |
|---|---|---|---|
| ALP (U/L) | -129.9 ± 14.6 | -14.4 ± 14.7 | -115.5 ± 13.2 |
| GGT (U/L) | -176.7 ± 25.6 | 6.7 ± 25.6 | -183.4 ± 23.3 |
| ALT (U/L) | -25.3 ± 3.4 | -5 ± 3.3 | -20.4 ± 3.1 |
| AST (U/L) | -15 ± 4.3 | 1 ± 4.2 | -16 ± 3.9 |
a16 patients (7%) were intolerant and did not receive concomitant UDCA: 6 patients (8%) in the OCALIVA 10 mg arm, 5 patients (7%) in the OCALIVA 5 mg→10 mg titration arm, and 5 patients (7%) in the placebo arm.1
ALP, alkaline phosphatase; ALT, alanine transaminase; AST, aspartate aminotransferase; GGT, gamma-glutamyl transferase; kPa, kilopascal; LS, liver stiffness; LSM, liver stiffness measurement; OLE, open-label extension; SE, standard error; TE, transient elastography; UDCA, ursodeoxycholic acid; ULN, upper limit of normal.
References
- OCALIVA full prescribing information. Morristown, NJ: Intercept Pharmaceuticals, Inc; 2022.
- Data on file: INT-PB-MED-00009.
- Trauner M, Nevens F, Shiffman M, et al. Long-term efficacy and safety of obeticholic acid for patients with primary biliary cholangitis: 3-year results of an international open-label extension study. Lancet Gastroenterol Hepatol. 2019;4(6):445-453. doi:10.1016/S2468-1253(19)30094-9
- Data on file: US-PB-MED-00899.
- Nevens F, Andreone P, Mazzella G, et al; for the POISE Study Group. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med. 2016;375(7):631-643. doi:10.1056/NEJMoa1509840
- European Association for the Study of the Liver. EASL clinical practice guidelines: the diagnosis and management of patients with primary biliary cholangitis. J Hepatol. 2017;67(1):145-172. doi:1016/j.jhep.2017.03.022