For adults with primary biliary cholangitis (PBC)1
STUDY DESIGN
STUDY DESIGN & PRIMARY ENDPOINT
OCALIVA was studied in POISE: the only clinical trial with 6 years of treatment follow-up in PBC2
Baseline characteristics:
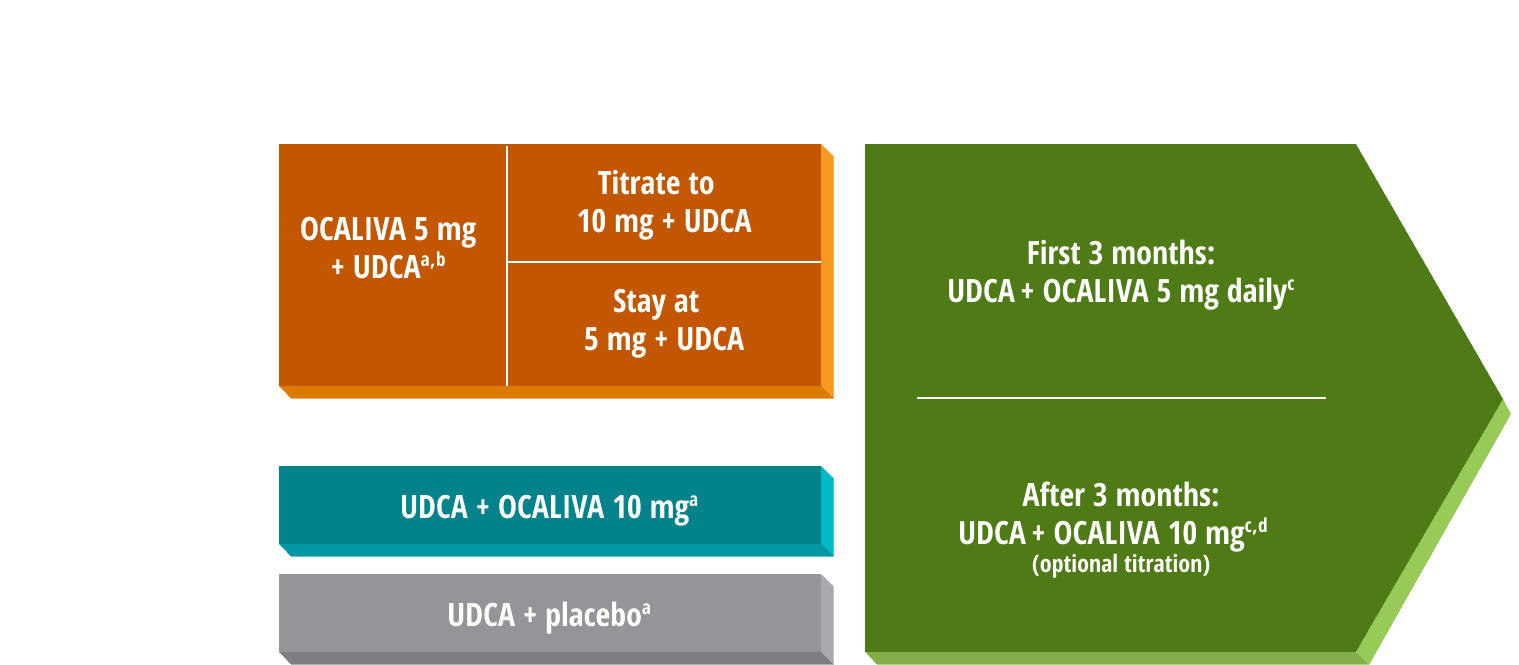
12-month clinical study
Adults with PBC (ages 29 to 86, mean 56) who were intolerant to UDCA or had an inadequate response to UDCA after at least 12 months, defined as (1) ALP ≥1.67x ULN and/or (2) bilirubin >1x ULN, but <2x ULN. If intolerant to UDCA, there was no UDCA use for ≥3 months (n=16).1,a
5-year OLE
An extension of the 12-month clinical study (n=193, being on a stable UDCA dose or being unable to tolerate UDCA and ALP ≥1.67x ULN and/or total bilirubin above ULN but <2x ULN). All patients received OCALIVA 5 mg for 3 months before optional titration based on response and tolerability.1,3
POISE Study Over 6 Years
Primary composite endpoint1,3:
- ALP <1.67x ULN
- ALP decrease of ≥15%
- Total bilirubin ≤ULN
Study limitations: In this OLE, no placebo or other comparators were included, and therefore no clinical conclusions should be made.4
Note: The 10 mg arm studied in POISE is not representative of the current
recommended dose schedule for OCALIVA.1,3
a16 patients (7%) were intolerant and did not receive concomitant UDCA: 6 patients (8%) in the OCALIVA 10 mg arm, 5 patients (7%) in the OCALIVA 5 mg→10 mg titration arm, and 5 patients (7%) in the placebo arm.1
bIn the 5 mg→10 mg titration group, 36 patients stayed at 5 mg, and 33 were titrated to 10 mg after 6 months.5
cAmong patients who entered the open-label extension, 13 (7%) were intolerant and did not receive concomitant UDCA during double-blind or open-label treatment with OCALIVA.1,3
dProtocol initially allowed doses up to 25 mg but was later amended to a maximum daily dose of OCALIVA 10 mg to ensure dosing per the approved label.3
ALP, alkaline phosphatase; ALT, alanine transaminase; AST, aspartate aminotransferase; GGT, gamma-glutamyl transferase; kPa, kilopascal; LS, liver stiffness; LSM, liver stiffness measurement; OLE, open-label extension; SE, standard error; TE, transient elastography; UDCA, ursodeoxycholic acid; ULN, upper limit of normal.
References
- OCALIVA full prescribing information. Morristown, NJ: Intercept Pharmaceuticals, Inc; 2022.
- Data on file: INT-PB-MED-00009.
- Trauner M, Nevens F, Shiffman M, et al. Long-term efficacy and safety of obeticholic acid for patients with primary biliary cholangitis: 3-year results of an international open-label extension study. Lancet Gastroenterol Hepatol. 2019;4(6):445-453. doi:10.1016/S2468-1253(19)30094-9
- Data on file: US-PB-MED-00899.
- Nevens F, Andreone P, Mazzella G, et al; for the POISE Study Group. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med. 2016;375(7):631-643. doi:10.1056/NEJMoa1509840
- European Association for the Study of the Liver. EASL clinical practice guidelines: the diagnosis and management of patients with primary biliary cholangitis. J Hepatol. 2017;67(1):145-172. doi:1016/j.jhep.2017.03.022