For adults with primary biliary cholangitis (PBC)1
LONG-TERM DATA
Data from POISE Open-Label Extension Study
Most patients receiving OCALIVA during the OLE met the composite endpoint2,3
Exploratory Analysis: Composite Endpoint in the OLE2,3,a

As in the double-blind phase, response was defined as meeting the following 3 criteria: ALP <1.67x ULN, ALP decrease of ≥15% from baseline, and total bilirubin ≤ULN.1,3
Study limitations: A maximum dose of OCALIVA 10 mg is presented, excluding data points as subjects titrated to greater than 10 mg daily.2 As this was a post hoc analysis, these results are exploratory, and no clinical conclusions should be made.
aOCALIVA baseline: For subjects who received placebo in the double-blind phase, baseline is the last assessment prior to the first OCALIVA dose in the long-term safety extension. For subjects who received OCALIVA in the double-blind phase, baseline is the mean of all available evaluations prior to double-blind treatment.2
bIncludes only patients who received OCALIVA in the double-blind phase.
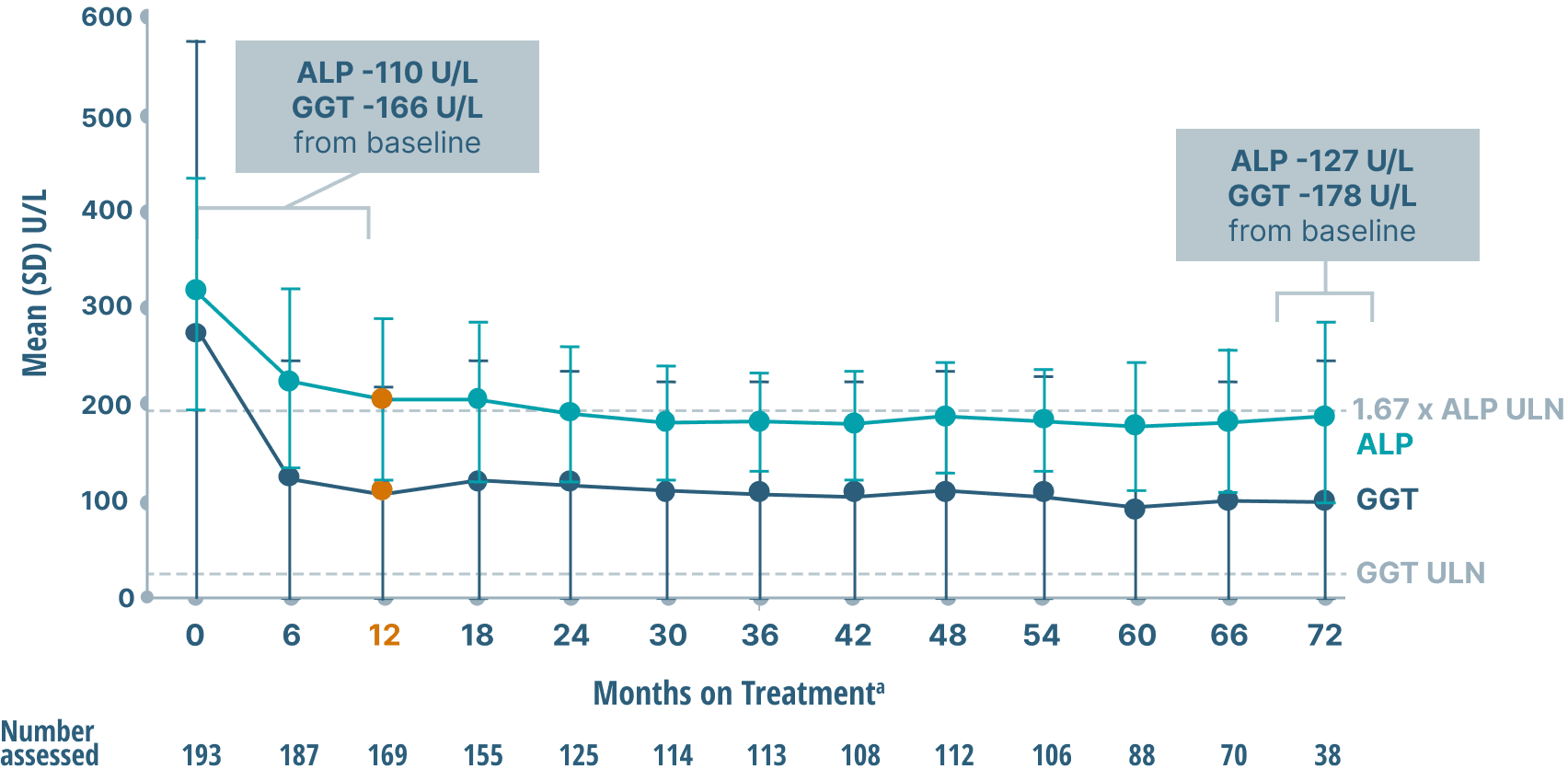
ALP, GGT, and bilirubin (key markers of cholestasis)
data over 6 years2,6
Mean ALP and GGT Levels
Mean Bilirubin Levels
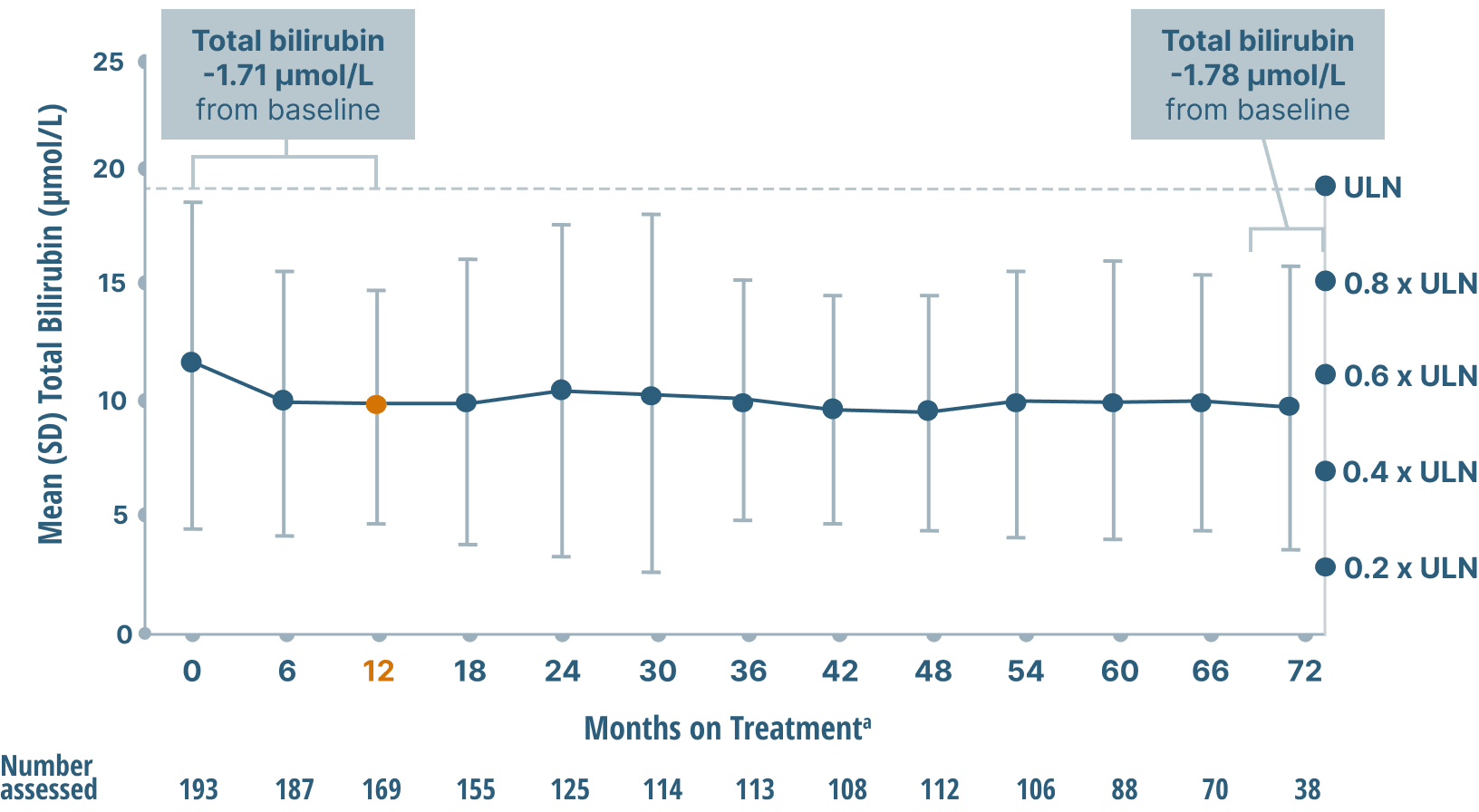
Study limitations: A maximum dose of OCALIVA 10 mg is presented, excluding data points as subjects titrated to greater than 10 mg daily.2 As this was a post hoc analysis, these results are exploratory, and no clinical conclusions should be made.
aOCALIVA baseline: For subjects who received placebo in the double-blind phase, baseline is the last assessment prior to the first OCALIVA dose in the long-term safety extension. For subjects who received OCALIVA in the double-blind phase, baseline is the mean of all available evaluations prior to double-blind treatment.2
ALT and AST (key markers of inflammation)
data over 6 years2
Mean ALT and AST Levels
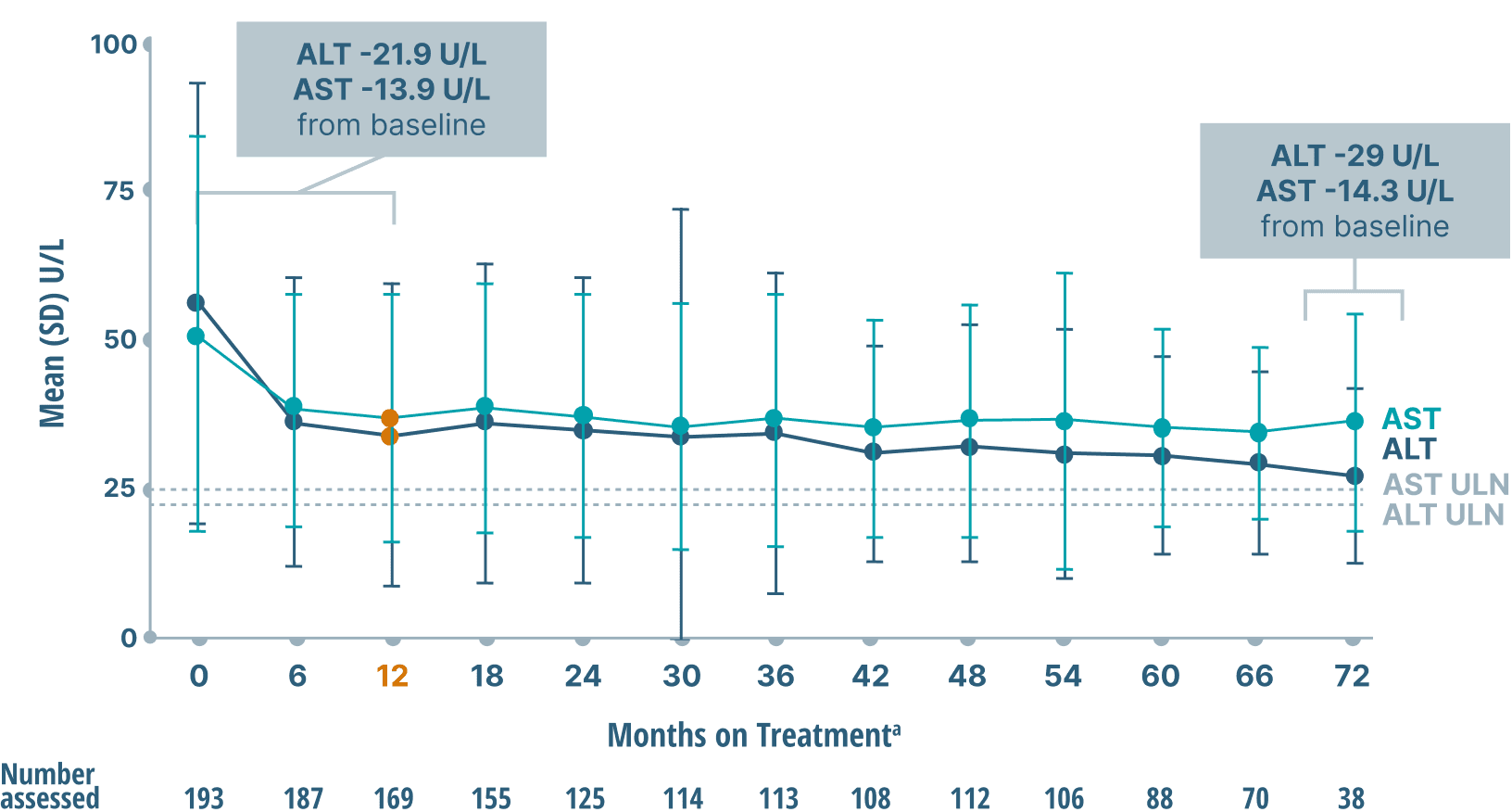
Study limitations: A maximum dose of OCALIVA 10 mg is presented, excluding data points as subjects titrated to greater than 10 mg daily.2 As this was a post hoc analysis, these results are exploratory, and no clinical conclusions should be made.
aOCALIVA baseline: For subjects who received placebo in the double-blind phase, baseline is the last assessment prior to the first OCALIVA dose in the long-term safety extension. For subjects who received OCALIVA in the double-blind phase, baseline is the mean of all available evaluations prior to double-blind treatment.2
Mean liver stiffness (AN INDICATOR OF FIBROSIS)
DATA OVER 6 years4
Mean LSM by TRANSIENT ELASTOGRAPHY (TE)
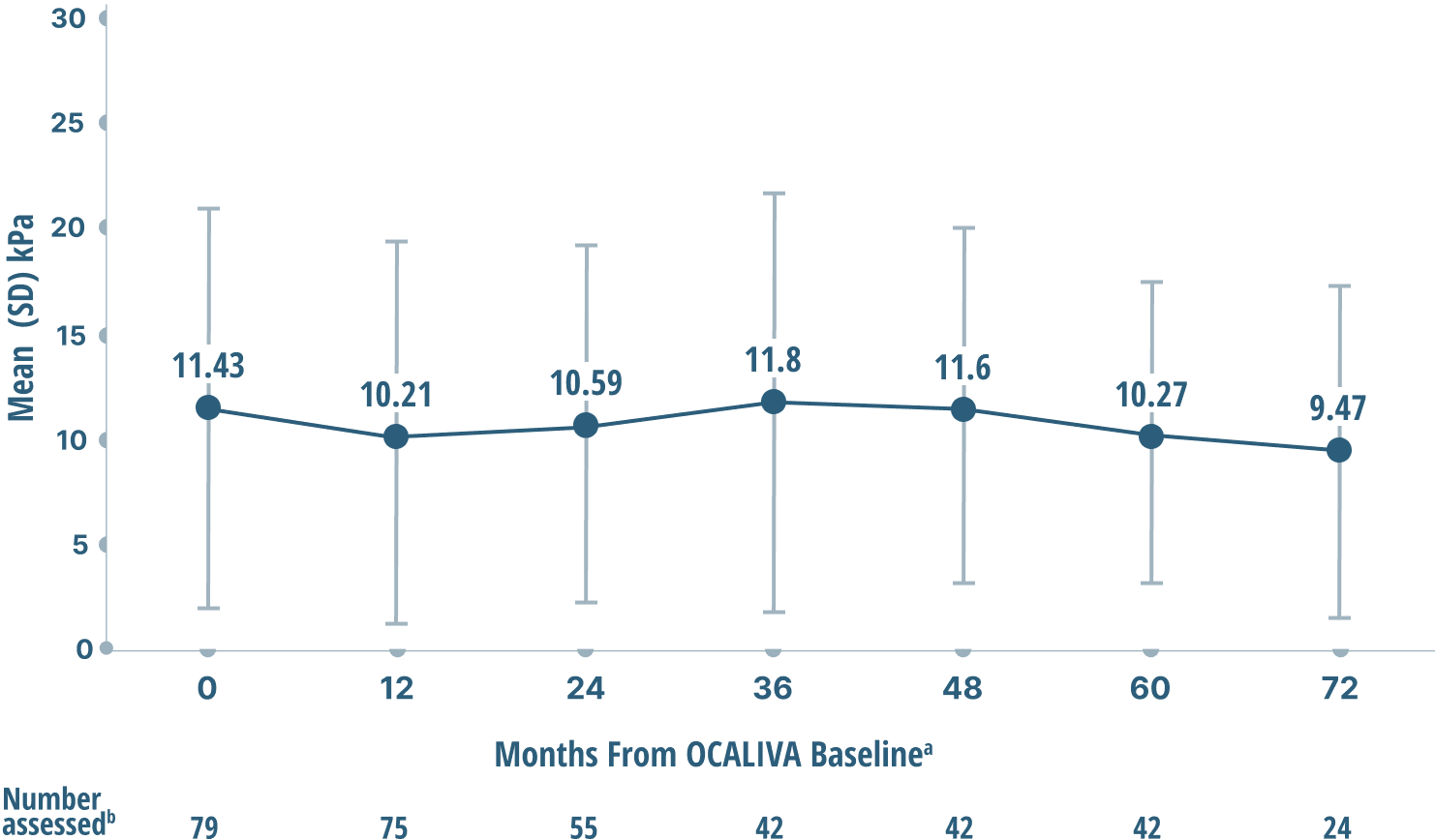
Study limitations: A maximum dose of OCALIVA 10 mg is presented, excluding data points as subjects titrated to greater than 10 mg daily.4 As this was a post hoc analysis, these results are exploratory, and no clinical conclusions should be made.
aOCALIVA baseline: For subjects who received placebo in the double-blind phase, baseline is the last assessment prior to the first OCALIVA dose in the long-term safety extension. For subjects who received OCALIVA in the double-blind phase, baseline is the mean of all available evaluations prior to double-blind treatment.2
bData were excluded if: 1) IQR/median exceeded 30%, or 2) transducers between baseline and follow-up did not match, or 3) the number of valid measurements were <10, >20, or N/A, or 4) measurements were not actually TE.4
ALP, alkaline phosphatase; ALT, alanine transaminase; AST, aspartate aminotransferase; GGT, gamma-glutamyl transferase; kPa, kilopascal; LS, liver stiffness; LSM, liver stiffness measurement; OLE, open-label extension; SE, standard error; TE, transient elastography; UDCA, ursodeoxycholic acid; ULN, upper limit of normal.
References
- OCALIVA full prescribing information. Morristown, NJ: Intercept Pharmaceuticals, Inc; 2022.
- Data on file: INT-PB-MED-00009.
- Trauner M, Nevens F, Shiffman M, et al. Long-term efficacy and safety of obeticholic acid for patients with primary biliary cholangitis: 3-year results of an international open-label extension study. Lancet Gastroenterol Hepatol. 2019;4(6):445-453. doi:10.1016/S2468-1253(19)30094-9
- Data on file: US-PB-MED-00899.
- Nevens F, Andreone P, Mazzella G, et al; for the POISE Study Group. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med. 2016;375(7):631-643. doi:10.1056/NEJMoa1509840
- European Association for the Study of the Liver. EASL clinical practice guidelines: the diagnosis and management of patients with primary biliary cholangitis. J Hepatol. 2017;67(1):145-172. doi:1016/j.jhep.2017.03.022